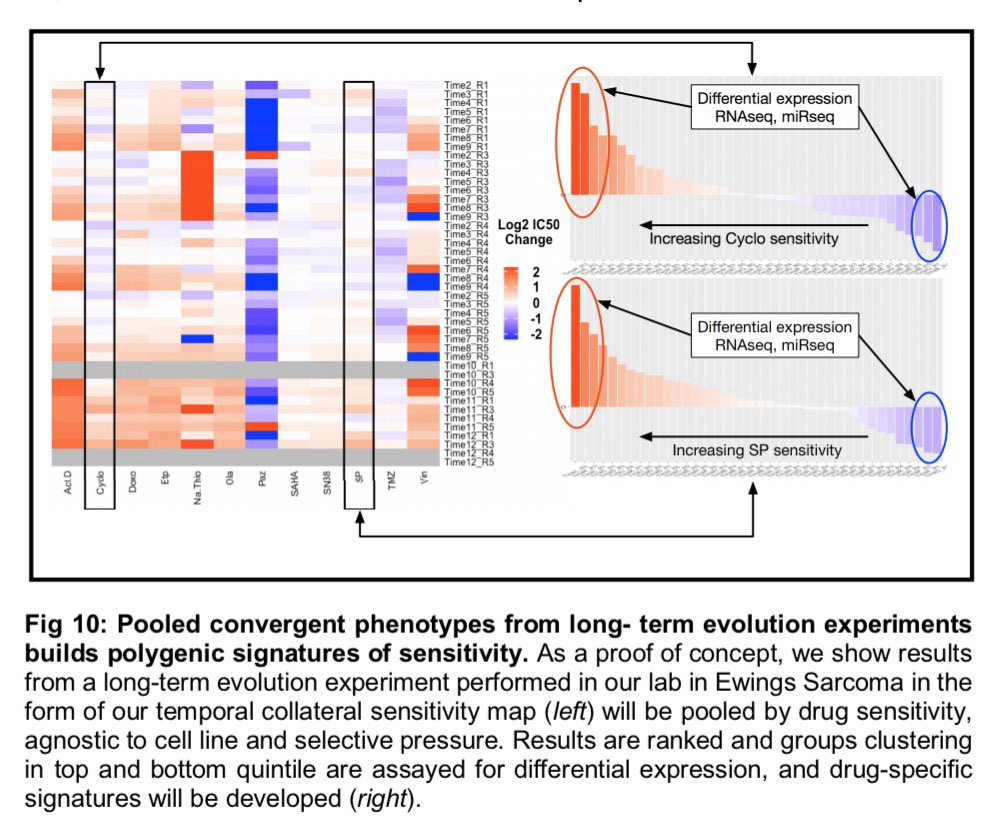
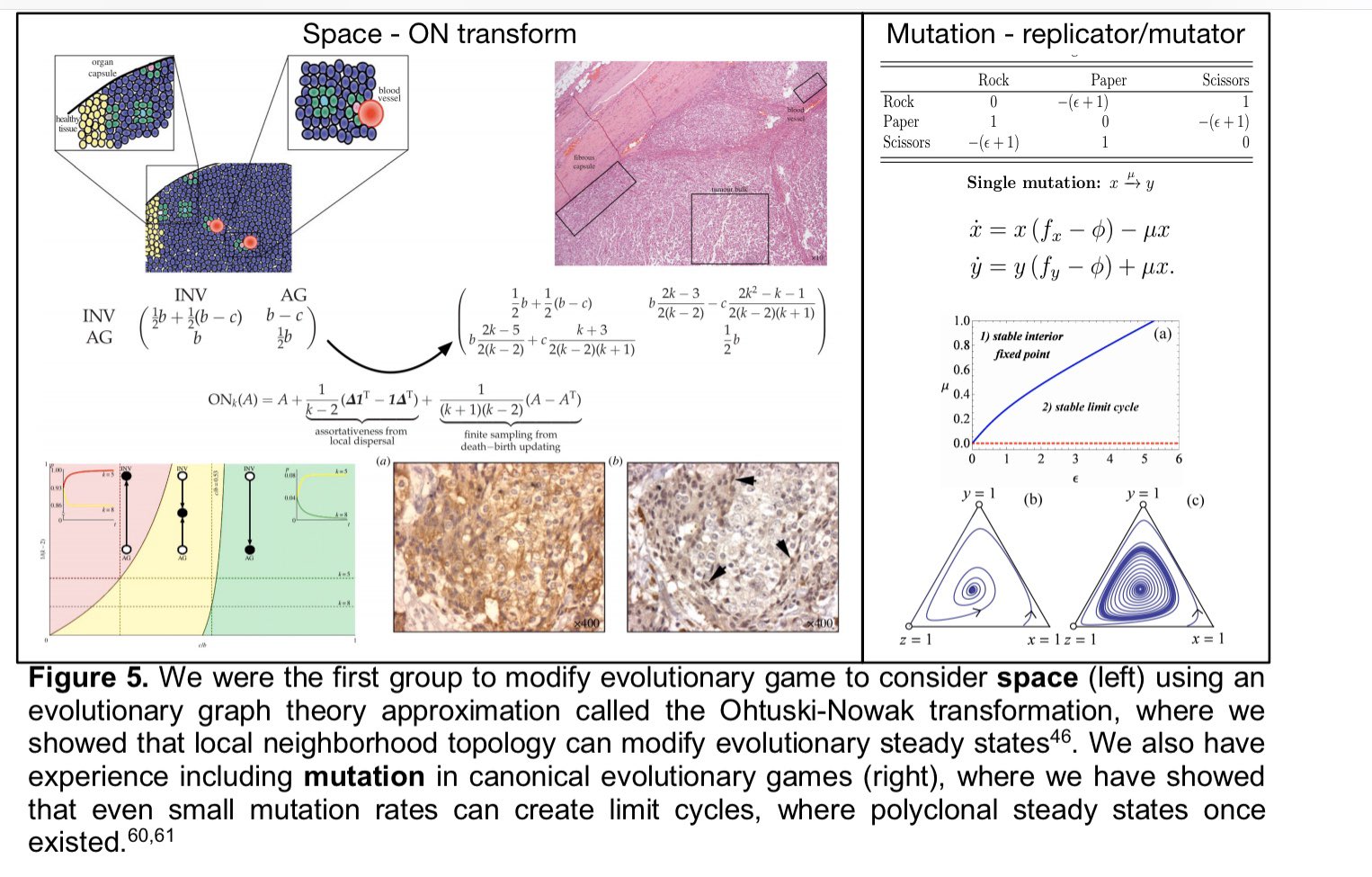
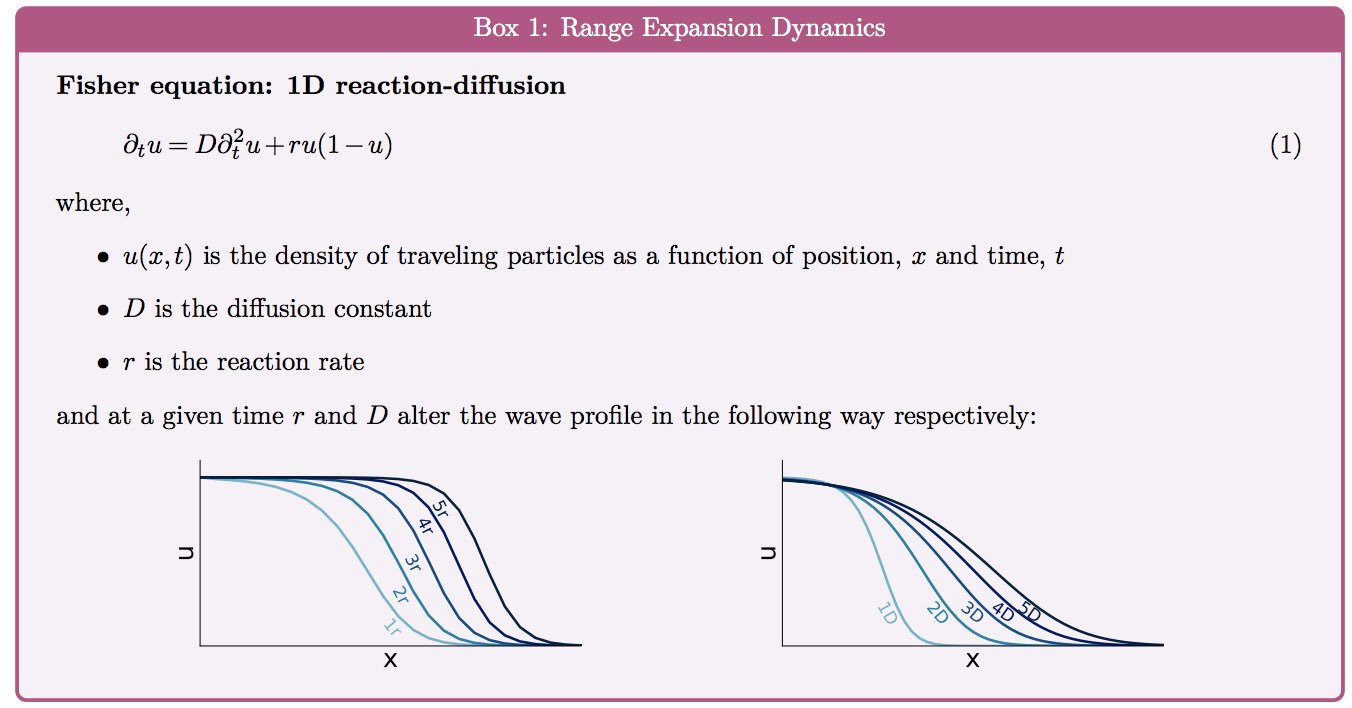
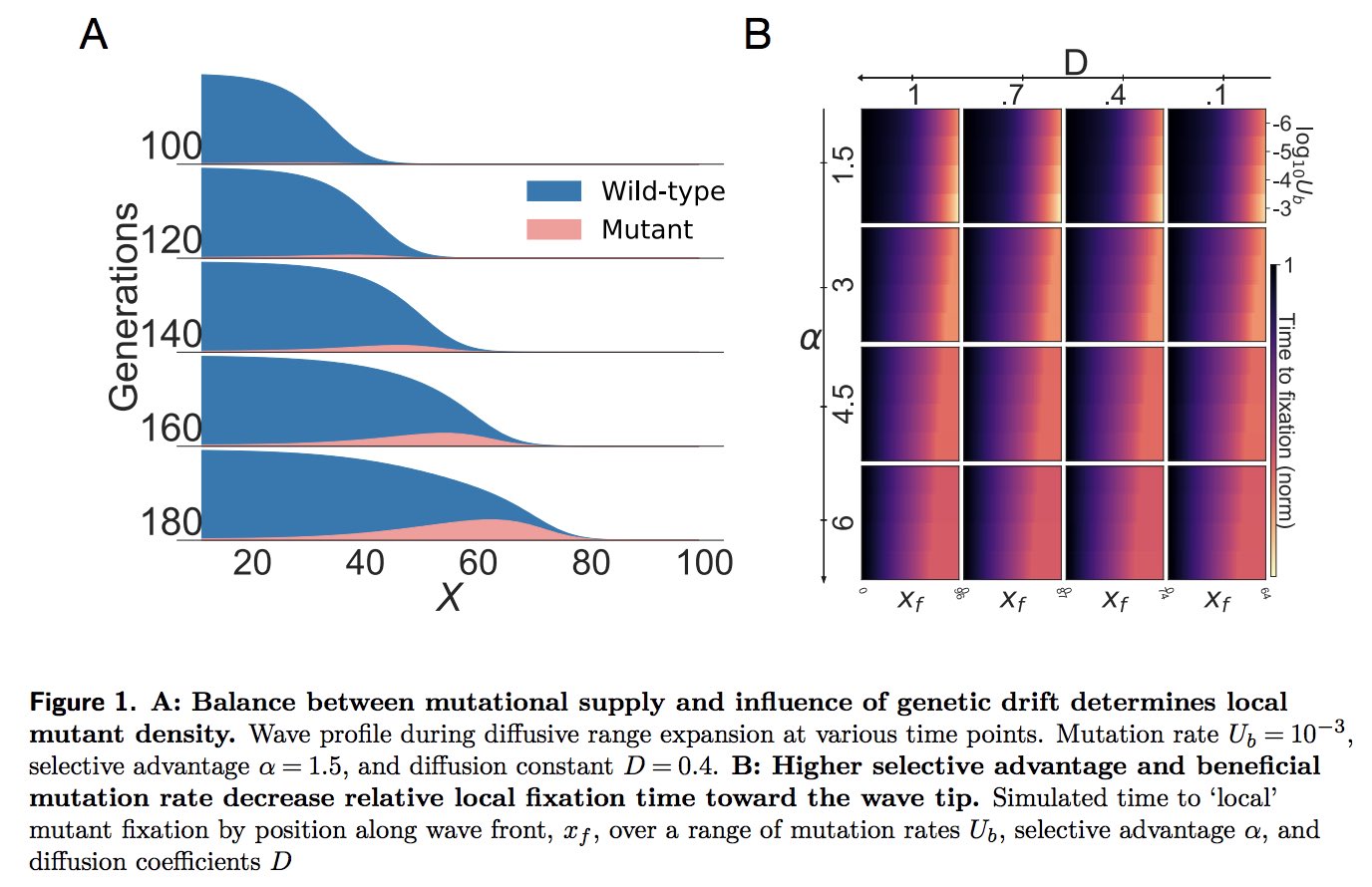
|
| 4 new post-docs have joined us in 2020 whose skills have a perfect amount of overlap to allow collaboration, but disparate enough so that we will constantly be learning from one another. Look out cancer and pathogens, we're coming for you. |
These three young scientists (along with some talented students who deserve their own post, but this one is about post-docs) really kick-started the work here in Theory Division, and helped spread the word about our work. When I finally got some real funding, and was able to hire more than one post-doc at a time, I started looking to hire two, as I knew Dr. Dolson was leaving us for the greener pastures of a tenure track job. This funny thing seems to happen to me... once I realize a post-doc is leaving I start to get really nervous. I think: who in their right mind would come work with me? How can I possibly ever replace (post-doc X) who is leaving?
These two questions are both ill-posed, but for different reasons:
1 - you can't replace (post-doc X), they are wonderful and unique in their own way, and 'replacing them' would stifle the growth of the lab anyways -- one needs new ideas, new skills, new faces...
2 - 'who would ever work with me??' is an impostor syndrome laden question -- but impostor syndrome is real, I ask myself this question every time (and still don't know the answer!)
So, I was shocked when Kyle Card, finishing his Phd in the Lenski lab, responded positively to my twitter DM asking if he was thinking about post-docs yet (as I had just read his excellent new paper: Historical contingency in the evolution of antibiotic resistance after decades of relaxed selection).

The summer before all this happened, I was in Cambridge lecturing to the PhD students in a unique program called CONTRA: Computational ONcology TRaining Alliance Innovative Training Network.
After my lecture, I was approached after my talk (which you can watch here) by Steph Owen, a talented PhD student working in cancer who was eager to get back to their roots in Physics. As a physicist myself was now doing cancer research, we hit if off immediately. The timing was such however, that Steph's PhD hadn't finished, and so we postponed the discussion of future work for a few months... which put our renewed conversation right at the beginning of COVID lock downs. Some of Steph's work can be read here: Combining measures of immune infiltration shows additive effect on survival prediction in high-grade serous ovarian carcinoma.At this point there were three people interested when I imagined I wouldn't find any, and now I was nervous about how to make this happen (strictly from a funding perspective). Then, the universe spoke. Nikhil Krishnan, a fourth year CWRU medical student in our lab (to whom I had promised PhD funding), got a Gates Fellowship to study Physics at Cambridge with Diana Fusco -- freeing up the funds to hire another person -- we'll call them the Nikhil Krishnan Post-doctoral fellow :)
The final piece of the puzzle I didn't know I was building was an unlooked for applicant. A friend and colleague, Kevin Wood from UM Biophysics visited us (actually the same weekend Kyle Card interviewed) to give a Biophysics colloquium, hosted by me and Mike Hinczewski (a friend and collaborator in CWRU Physics). A grad student in his lab (essentially Black Mirror version of our own lab), Jeff Maltas, was just finishing his PhD. It turns out that we had reviewed half of his thesis papers (anonymously) so I was quite familiar with his (excellent!) work (I take some credit for Figure 6 :) in: Pervasive and diverse collateral sensitivity profiles inform optimal strategies to limit antibiotic resistance). I didn't think Jeff would want to work with us, but invited him for a visit anyways. and then, there were 4...
I couldn't have possibly decided between the 4, so I decided to go with them all, and sort the funding out later. Turns out, they did it for me -- between a computational genomics T32 and a competitive NIH diversity supplement, the universe (and my talented new young scientists) provided, and I am SO EXCITED to see what science we do together. There is just enough overlap between us all that we can speak freely and in our own scientific mother tongues, and enough separation that each is synergistic with the others. I feel incredibly honored to be able to host these young scientists and can't wait to see what science we're able to in Theory Division in 2021.